DLPGEN: Preparing Molecular Dynamics Simulations with Support for Polarizable Force Fields
Abstract
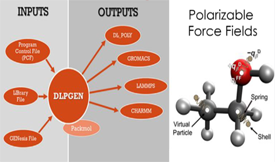
Preparing input files for molecular dynamics (MD) simulations can be a tedious task, particularly if different MD programs need to be used. Most simulation packages are accompanied by applications that handle this task, and, in some cases, software to perform interconversion between MD programs can be found. However, the conversion between different types of files is not always foolproof or the force field may not be fully supported, as quite often observed with polarization models. This work describes the program DLPGEN, which produces input files for the MD programs GROMACS, CHARMM, DL_POLY, and LAMMPS. The program can prepare polarizable force fields using a self-consistent field approach or with a dual thermostat-extended Lagrangian model. For GROMACS, a new polarization scheme suitable for the simulation of molecules containing more than one virtual particle is described. Results obtained for ethanol in the liquid state revealed that the system configurational energy, liquid density, and self-diffusion coefficient obtained with GROMACS are in good agreement with the data found with LAMMPS and CHARMM. In the case of DL_POLY, a problem with the Shells temperature control was found, suggesting that this program may not be suitable for the simulation of molecules containing multiple Drude particles.
Return Previous Next