Laboratórios e batas brancas. Destiladores, tubos de ensaio, buretas e provetas. Ácidos, bases e sais. Fumarolas, cores e odores. Sínteses e análises. Tintas, vernizes, detergentes e plásticos. Alimentos e medicamentos. Células biológicas, vírus e bactérias. Eléctrodos, baterias e sensores. Fórmulas e modelos moleculares. Arte e geometria. Átomos e moléculas em movimento. Indústrias química, agroquímica e farmacêutica. Cisões e fusões nucleares. Estrelas, cometas, e planetas. Internet, semi-condutores e computadores. Agatha Christie e Conan Doyle.
Estas são realidades onde a Química está sempre presente. A sua vasta área é de importância crucial desde a escala atómica à galáctica, contribuindo para desvendar os mecanismos da Natureza, e para a saúde, bem-estar e progresso do ecosistema.
Contudo, em alguns meios sociais ainda surge, por vezes, a ideia de que os químicos andam nos laboratórios a misturar reagentes e a usar computadores, em lucubrações teóricas e tentativas de descobrir o “elixir da longa vida”, ou a inventar bombas e outros produtos letais que levarão à destruição da Humanidade.
Esta imagem alquímica, guerreira e poluente é irreal. Urge rebatê-la, combatendo a subjacente iliteracia científica.
Os cientistas são, em geral, ética e profissionalmente responsáveis, embora muitas vezes ultrapassados por decisões políticas irreflectidas ou ambiciosas. Qualquer actividade científico-tecnológica que busque a inovação pode ter o “reverso da medalha”. Contudo, somente os cientistas e técnicos, com os seus conhecimentos e métodos, podem corrigir consequências menos positivas que eventualmente surjam. Investir em ciência e tecnologia (e humanidades, sublinhe-se), com critérios justificados e dando efectivas oportunidades às gerações mais novas, é o caminho para o progresso saudável da Sociedade.
Neste artigo (uma contribuição para as celebrações do Ano Internacional da Química, AIQ2011) divulgam-se aspectos essenciais da Química Computacional, um ramo interdisciplinar da Química que serve todas as suas especialidades, tecnologias e indústrias afins. Os alicerces foram lançados nos anos 50 do século passado, em simultâneo com a implementação dos primeiros computadores. Actualmente, qualquer pessoa, curiosa e persistente, pode aprendê-la servindo-se dum vulgar computador pessoal ligado à Internet. É, realmente, um desafio. Até há quem diga que a Química Computacional é um incomensurável tronco pertencente aos séculos XXI e vindouros, do qual alguns ramitos começaram a cair no século XX.
Objectivos, Métodos e Programas
No que se segue, os leitores menos familiarizados com estes temas poderão encontrar termos e técnicas que desconhecem. Assim, recomenda-se a consulta das referências [1-6]. São fontes de material didáctico, software e técnicas de programação que cobrem grande parte dos tópicos abordados neste artigo. Adicionalmente, nos endereços dos grupos de investigação, mencionados ao longo do texto, encontram-se outras referências a assuntos das respectivas especialidades. A Química Computacional consta, em última instância, do desenvolvimento e utilização de software dedicado à resolução de problemas químicos, bioquímicos, tecnológicos e industriais. A sua vastidão leva a que se identifiquem algumas vertentes como a Modelação Molecular (ou Simulação Molecular), Quimiometria, Quimioinformática e Bioinformática que, embora com notáveis intersecções, sistematizam objectivos e métodos específicos:
As exigências de cálculo numérico e simbólico, e de arquivo, edição e visualização de enormes quantidades de informação química, tornam o computador instrumento-chave em Química Computacional. Contudo, tratando-se duma máquina, tem de ser instruído. A programação surge, assim, como um recurso imprescindível, em liguagens como o Fortran, C++, Java e Prolog. Actualmente, existem vários pacotes de software, académicos ou comerciais, com visualização molecular interactiva (Gaussian, Gromacs, Towhee, Dock, Maddam, Spinus, ChemOffice, etc.) que podem ser utilizados quase como “caixas-pretas” e que servem proficuamente as diversas vertentes da Química Computacional. No entanto, os grupos de investigação têm frequentemente de desenvolver os seus próprios programas, quando o tipo de problemas não está contemplado nos pacotes ou estes não apresentem a fiabilidade requerida. A programação é um desafio e um prazer intelectual (experimente-se!) e, saliente-se, que são químicos, bioquímicos, físicos e biólogos que mais contribuem para esses softwares, cedidos muitas vezes gratuitamente.
Há que referir dois tipos essenciais de programas computacionais conforme os problemas a resolver:
Por exemplo, a computação da estrutura electrónica duma molécula ou a dinâmica molecular dum líquido são problemas em geral bem definidos, quer nos dados a introduzir quer nos resultados expectáveis. Os respectivos programas são do tipo convencional, isto é, um conjunto de operações elementares e sequenciais que processam dados completos e precisos, conduzindo a respostas únicas. Baseiam-se fortemente em métodos matemático-numéricos, mais ou menos sofisticados, recorrendo ao cada vez maior poder de cálculo dos computadores.
Contudo, existem outros problemas químicos que não podem ser eficientemente resolvidos através daqueles programas. O aspecto essencial não é mais poder computacional e sofisticação matemática, mas uma aproximação diferente. Considere-se, por exemplo, a determinação do número de isómeros da molécula C26H54O que são álcoois, o planeamento de sínteses químicas com vista a novos medicamentos e materiais, a estereoquímica de proteínas e a determinação das conformações de energia mínima de outras moléculas complexas, as sequências genéticas, a selecção de parâmetros óptimos em análise instrumental, o controlo automático de unidades industriais de larga escala, a dispersão de poluentes, e a análise e tratamento de bases de dados químicas. Estes casos tipificam a denominada explosão combinatória, ou seja, o número de hipóteses e detalhes é tão grande que não é possível utilizar apenas programas convencionais para obter respostas em tempo útil, mesmo utilizando a força bruta dum supercomputador e técnicas numéricas sofisticadas. Por outro lado, quer os dados quer os resultados destes casos possuem geralmente incertezas intrínsecas implicando que as operações adequadas para a sua resolução não tenham uma sequência bem definida (como nos algoritmos convencionais), e a possibilidade de várias respostas com diferentes níveis de confiança. A abordagem destes problemas recorre a métodos de Inteligência Artificial, os quais tentam simular num computador o raciocínio inteligente, os mecanismos genéticos, e a aquisição e manipulação do conhecimento. Uma das aproximações-chave baseia-se no facto do cérebro, constituído por uma rede de neurónios massivamente paralela, não funcionar sequencialmente. Os neurónios têm, individualmente, um funcionamento semelhante e porventura simples, mas dispostos em arquitecturas paralelas possuem a capacidade de processamento simultâneo e integrado, trocando entre eles a informação recebida do exterior ou gerada internamente, e manifestando, entre outros, dois aspectos essenciais do raciocínio: os saltos intuitivos e a capacidade de aprendizagem com a experiência.
Quem faça palavras cruzadas reconhecerá os saltos intuitivos: certamente não executa a procura exaustiva de todas as palavras que conhece e que possam caber no espaço indicado; basta muitas vezes uma única letra para ter o seu “salto”. Um algoritmo convencional não teria outra alternativa senão a da procura exaustiva. Por outro lado, só se aumenta o desempenho da condução automóvel (isto é, só se aprende) com a experiência repetida e a memorização. Um programa convencional não é capaz de memorizar e aprender. Uma vez escrito fica imutável até que o programador o altere.
Os programas de Inteligência Artificial (redes neuronais, algoritmos genéticos, “expert systems”, etc.) reproduzem num computador essas capacidades com considerável sucesso, suportando a resolução de problemas de larga escala e complexidade. Por exemplo, o número de moléculas que potencialmente podem actuar como fármacos estima-se ser da ordem de 1040. Deste número astronómico de possibilidades apenas uma infíma fracção será eventualmente sintetizada, e desta somente um número ainda menor será submetido a "high throughput screening" para testes humanos. Qualquer procedimento computacional concebido para esse fim deve ser capaz de considerar o número de potenciais fármacos sem efectivamente inspecionar cada uma das estruturas moleculares.
Esses programas tornam possível, também, a incorporação duma área de crescente interesse químico e tecnológico: a lógica difusa ("fuzzy logic"). Esta lógica mostra que, afinal, as regras formais de raciocínio estabelecidas por Aristóteles são demasiadamente rígidas, não permitindo o tratamento adequado de inúmeros problemas complexos. De facto, a resposta a uma questão pode não ser exactamente “sim” ou “não”, mas “talvez”; a cor dum objecto pode não ser exactamente “preto” ou “branco”, mas “cinzento”, existindo dentro deste uma infinidade de tonalidades. Actualmente, é vulgar encontrar processadores “fuzzy” em máquinas de lavar roupa e louça, automóveis e ar condicionado. Estes processadores, contrariamente aos digitais (de 0’s ou 1’s), permitem uma variação contínua entre 0 e 1, conduzindo a controlos suaves com substanciais poupanças de energia e água.
Em grupos de investigação de universidades, institutos e empresas portuguesas desenvolve-se uma intensa e frutuosa actividade nas diferentes vertentes da Química Computacional. No que se segue, selecionaram-se aplicações desenvolvidas nalguns desses grupos, representativas da importância deste ramo da Química. Sublinhe-se, no entanto, que, neste artigo, não se pretende fazer uma revisão da actividade científica nacional nesta área, mas apenas exemplificar uns quantos aspectos computacionais da química fundamental e aplicada, bioquímica, agroquímica e medicina.
Numa publicação posterior será apresentada uma revisão exaustiva dos grupos de Química Computacional em Portugal, resultante de um inquérito realizado a nível da Sociedade Portuguesa de Química. Isso seria inadequado neste artigo considerando o número de grupos cuja actividade se estende do Minho ao Algarve, Madeira e Açores. Assim, os grupos não mencionados na seleção seguinte serão certamente referidos na revisão, a qual esperamos que seja também apresentada numa página da Internet com vista a uma ligação direta aos endereços dos diferentes grupos e permanente actualização.
Nanoclusters e líquidos iónicos
O cloreto de potássio (KCl) é um sal (halogeneto alcalino) utilizado, por exemplo, em fertilizantes líquidos, processamento de alimentos e medicina. Cristaliza numa geometria cúbica e funde a 1043 K (770 ºC). As suas propriedades (como as de qualquer substância) são geralmente determinadas com amostras que por muito pequenas, sob o ponto de vista macroscópico, contêm sempre números de iões (K+ e Cl-) da ordem do número de Avogadro (6,023x1023). Lord Kelvin deu a noção da grandeza deste número num modo apelativo: "Encha-se um copo com água cujas moléculas foram marcadas. Deite-se essa água num dos oceanos e deixe-se as moléculas espalharem-se por todos os mares do mundo. Encha-se de novo o copo em qualquer dos mares. Nele estarão cerca das 100 moléculas inicialmente marcadas”.
Como se comportarão microagregados de KCl com números de iões apenas da ordem das dezenas, centenas ou milhares? Apresentarão também transições e coexistência de fases? E a que temperaturas? O cloreto de potássio pode formar vidros quando o líquido é arrefecido bruscamente. E os microagregados? Serão as leis dos grandes números aplicáveis a sistemas com um número muito pequeno de moléculas? Note-se que a origem de qualquer material são microagregados com dimensões lineares da ordem do nanometro (10-9 m), designados por nanoclusters. Estas questões são de importância fundamental.
A propósito, Erwin Schrödinger, no famoso clássico "What is life?”, interroga-se sobre as regularidades observadas nas células biológicas, com fina reprodutibilidade, e consequentes do ADN que é constituído por um número de moléculas muitissimo inferior ao número de Avogadro. De facto, sob o ponto de vista estritamente macrofísico, apenas de sistemas macroscópicos deveriam emergir propriedades e leis com flutuações desprezáveis.
Estes aspectos são particularmente adequados para a simulação computacional pelo método da dinâmica molecular. Sucintamente, um programa de dinâmica molecular calcula as posições e velocidades das partículas em cada instante (ou seja as suas trajectórias) usando a lei de Newton (força = massa x aceleração). A partir das trajectórias o computador pode produzir filmes, e calcular propriedades como a temperatura, pressão, energias cinética e potencial, energias livres, coeficientes de difusão, viscosidade, condutividades térmica e eléctrica, correlações espaciais e temporais, e intensidades espectroscópicas.
Os resultados obtidos numa série de simulações sobre transições e coexistência de fases, ilustradas por filmes produzidos por Pedro Rodrigues, estão disponíveis em [7]. As simulações mostram que os nanoclusters apresentam transições de fase sólido-líquido cujos pontos de fusão aumentam com a dimensão dos clusters, tendendo para o valor da temperatura de fusão do macrosistema (os clusters com apenas 512 iões já têm pontos de fusão de ~1000K) e formam vidros quando o líquido é arrefecido bruscamente. A partir de ~1000 iões sustêm, também, coexistência de fases (fig. 1). Não serão, realmente, os nanoclusters os “genes” dos macro materiais?
Figura 1. Coexistência sólido-líquido num nanocluster de KCl com ~5000 iões
A respeito de materiais, Rui Fartaria [8], por exemplo, tem realizado simulações sobre nano-compósitos de polímeros argilosos, suspensões coloidais e cristais líquidos. Por outro lado, no CICECO [9], simulam-se diferentes materiais relacionados com investigações principalmente orientadas para a tecnologia.
No contexto de sistemas iónicos, os líquidos iónicos em particular, com aplicações importantes no processamento de celulose e combustíveis nucleares, em baterias, na extracção de compostos vegetais com fins nutricionais, cosméticos e farmacêuticos, e em química verde, têm tido contribuições computacionais de vários grupos como o de Canongia Lopes [10], em colaboração com grupos experimentais.
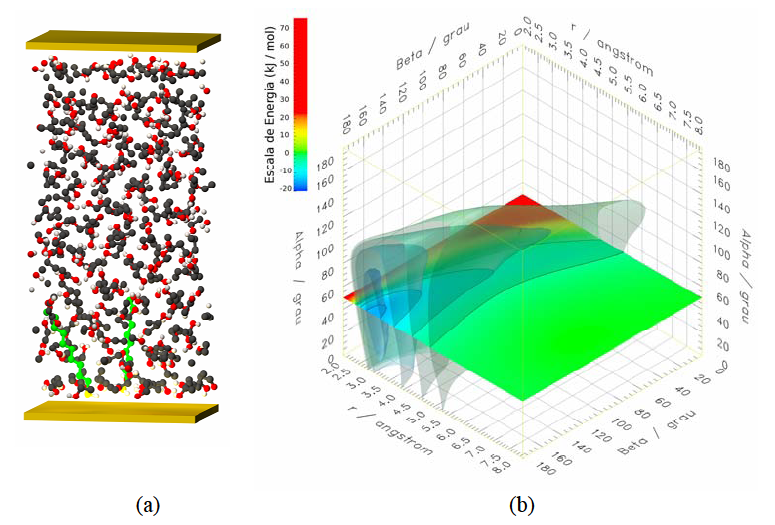
Os alcanotióis são facilmente adsorvidos por eléctrodos de ouro através do grupo tiol (o grupo tiol é –SH), dada a grande afinidade entre o enxofre e o ouro. Após a adsorção a partir de uma solução em etanol, as moléculas automontam-se sobre a superfície do ouro constituindo monocamadas densas e estáveis (fig. 2a). O controlo electroquímico deste processo permite a modificação química das caudas alquílicas com a introdução de outras moléculas, como as do fulereno C60, dando lugar, por exemplo, ao desenvolvimento de diferentes tipos de sensores. Por outro lado, a electro-oxidação de compostos fenólicos adsorvidos em eléctrodos de metais nobres é uma das técnicas possíveis para combater a poluição.
Figura 2. (a)um passo na adsorção e automontagem sobre eléctrodos de ouro de moléculas de decanotiol (a verde) dissolvidas em etanol; (b) superfície de energia potencial do etanol-ouro.
O grupo de Fernando Fernandes [11] tem realizado simulações computacionais dos mecanismos envolvidos nestes processos, em colaboração com grupos experimentais de electroquímica interfacial, utilizando uma conjugação de métodos quânticos e de Monte Carlo. Este método é, em muitos aspectos, equivalente ao de dinâmica molecular, mas em vez de calcular as trajectórias das partículas deterministicamente, segundo a lei de Newton, faz uma amostragem estocástica (um processo estocástico caracteriza uma evolução sujeita às leis da probabilidade) das posições e/ou das velocidades. Estes problemas, e outros do domínio das interações moleculares, reactividade e ciência dos materiais, têm tido contribuições de mais grupos como o de Ferreira Gomes [12].
Sublinhe-se que a Mecânica Quântica está sempre subjacente nos métodos da Química Computacional, embora muitas vezes de forma não explícta. Estruturas atómicas, moleculares e nucleares, espectroscopia, campos de forças aproximados para dinâmica molecular e Monte Carlo, e superfícies de energia potencial (fig. 2b) para o estudo de interfaces e reacções químicas, por exemplo, têm invariavelmente o suporte quântico. Neste contexto, o grupo de António Varandas [13] tem feito desenvolvimentos seminais da química quântica, e dos respetivos métodos computacionais, com diversas aplicações, em particular à química atmosférica. Entre outros, os grupos de Benedito Cabral [14] e de Prates Ramalho [15], também se têm dedicado à simulação quântica de matéria condensada (sólidos, fluídos e clusters).
Redes neuronais e previsão de espectros de RMN
A ressonância magnética nuclear (RMN) é uma técnica espectrométrica utilizada em todos os ramos da química, na indústria e em medicina. Fundamenta-se no facto dos protões e neutrões dos núcleos atómicos terem spins (grosseiramente, movimentos de rotação) que originam campos magnéticos intrínsecos os quais podem entrar em ressonância com campos magnéticos aplicados externamente. Os núcleos dos átomos duma molécula estão rodeados de electrões que exercem um efeito de blindagem aos campos externos aplicados. É este efeito que torna a RMN útil para a identificação de estruturas moleculares, uma vez que a blindagem depende da distribuição electrónica em torno de cada núcleo a qual determina as várias ligações químicas das moléculas. Assim, as frequências de ressonância nucleares são diferentes conforme as vizinhanças dos núcleos de uma dada molécula. Essas diferenças designam-se por desvios químicos, relativamente a um padrão, por exemplo, o tetrametilsilano (TMS). A técnica contempla vários núcleos como 1H (protão), 13C, 15N, 19F e 31P.
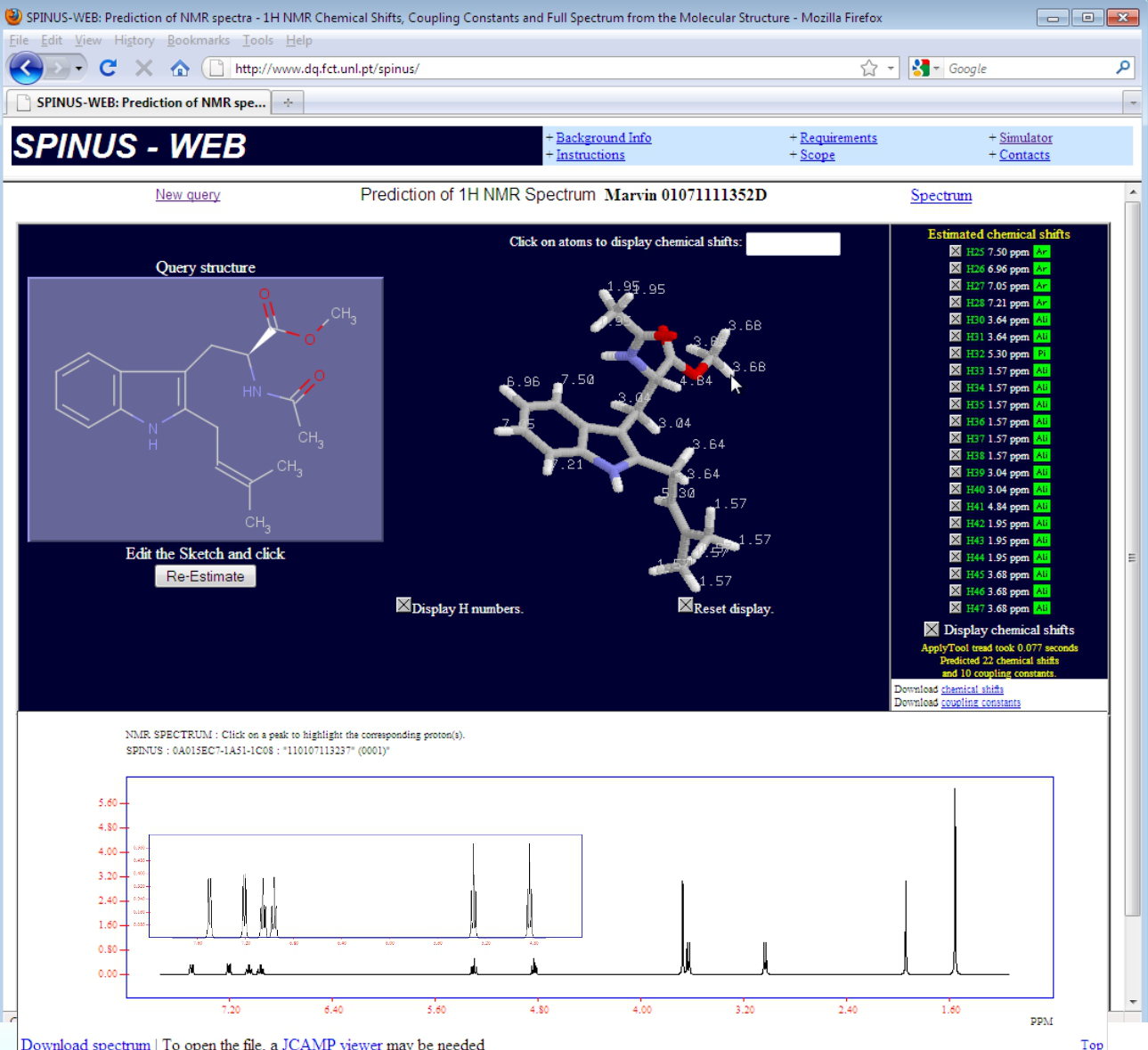
Quando se sintetiza uma molécula nova é necessário comprová-lo. Será então da maior importância prever antecipadamente o seu espectro, por exemplo de RMN, e depois compará-lo com o obtido experimentalmente. Tal é possível com programas de redes neuronais. Esses programas aprendem com exemplos de espectros de diferentes tipos de moléculas conhecidas. Uma vez ensinados podem prever, com grande fiabilidade, os espectros de moléculas novas. Por exemplo, o programa SPINUS, desenvolvido no grupo de Aires de Sousa [16] para espectros de RMN de protão, e disponível em [17]. Tudo o que o utilizador tem de fazer é desenhar a molécula que pretende com a interface gráfica incluída e, depois, com um simples clique obtem o espectro em termos de desvios químicos (fig. 3). Cada sinal do espectro corresponde a um átomo de hidrogénio do composto com vizinhanças diferentes. Além dos desvios químicos são também calculados os acoplamentos spin-spin de núcleos adjacentes, responsáveis pela estrutura fina dos espectros.
Figura 3. Interface do programa SPINUS com as estruturas de uma molécula (2D e 3D) e espectro de RMN de protão (com detalhes de estrutura fina)
Uma outra aplicação é na elucidação estrutural, ou seja, deduzir a estrutura para um composto novo a partir de dados espectroscópicos experimentais. Vários programas com este objectivo incorporam previsões de espectros, como as do SPINUS, para filtrar entre as diferentes estruturas-candidato possíveis.
O grupo referido tem desenvolvido, também, outras aplicações dos métodos de inteligência artificial, entre elas, a distribuição geográfica de amostras petrolíferas, classificação de reacções enzimáticas à escala genómica e, particularmente Diogo Latino [18], ajustes não paramétricos de superfícies de energia potencial intermolecular.
Dinâmica de proteínas, dendrímeros e lípidos
A visualização molecular é um recurso indispensável, em particular para moléculas cruciais nas estruturas celulares e processos biológicos. Complementa os cálculos, detecta pormenores que poderiam ficar escondidos e sugere novas experiências. Por exemplo:
A lisozima é uma proteína enzimática (existe na clara dos ovos de galinha, leite humano, saliva, etc.) fazendo parte do sistema imunitário natural. Sendo uma forma natural de protecção a bactérias como a Salmonella, E.coli e Pseudomonas, a sua deficiência na alimentação, em especial dos recém-nascidos, tem sido associada a bronco-pneumonias e diarreias.
As interacções entre os grupos funcionais do fosfolípido cardiolipina (CL) resultam em conformações moleculares que se sugerem ser determinantes para o bom funcionamento da membrana interna do mitocôndrio, estando a deficiência de CL associada ao síndrome de Barth.
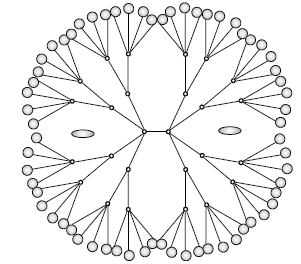
Os dendrímeros, são moléculas poliméricas cuja arquitectura é caracterizada por ramos sucessivos, por vezes aproximadamente esféricos, que lhes conferem as designações de dendrímeros de 1ª, 2ª, 3ª,... gerações. Existem diferentes famílias de dendrímeros que embora com a mesma arquitectura se distinguem, essencialmente, pelos tipos de monómeros (as unidades básicas dum polímero). Têm semelhanças com as proteínas na medida em que são moléculas poliméricas e, no caso dos dendrímeros peptídicos (uma família que tem tido um crescente desenvolvimento desde 2005), os respectivos monómeros são aminoácidos tal como nas proteínas. Relativamente a estas, é de destacar sua grande flexibilidade molecular. As aplicações dos dendrímeros são variadas e promissoras como em bioquímica, nanomedicina e indústria. A arquitectura e propriedades químicas destas moléculas torna-as particularmente adequadas para, por exemplo, encapsularem (fig. 4) fármacos e genes transportando-os, respectivamente, para tecidos cancerosos e ADN de células biológicas. Para além da delicada estratégia da técnica, ela possibilita a atenuação dos efeitos de citotoxicidade desses tratamentos.
Filmes, obtidos pelo método da dinâmica molecular, sobre a lisozima, a cardiolipina (em situações diferentes) e de um dendrímero peptídico (onde se observa a sua grande flexibilidade molecular relativamente às proteínas), produzidos pelo grupo de Miguel Machuqueiro, estão disponíveis em [19].
Figura 4. Esboço de uma “caixa” dentrítica de 3ª geração encapsulando duas moléculas
Outros grupos, como os de Cláudio Soares [20] e de António Baptista [21], têm contribuído também para a modelação e dinâmica de proteínas, dendrímeros e aspectos afins.
Actividade enzimática e concepção de fármacos
As enzimas são proteínas que catalizam reacções químicas A maioria dos processos biológicos necessita de enzimas cuja actividade é afectada por outras moléculas, as quais podem actuar como inibidores ou activadores. Muitos fármacos e venenos são inibidores enzimáticos.
Os mecanismos reaccionais, ou seja, o modo como as ligações químicas das moléculas se quebram e formam, dependem directamente da estrutura electrónica das moléculas. Um passo determinante de qualquer reacção é a formação do chamado complexo activado, isto é, um agregado entre as moléculas dos reagentes com conformações e orientações relativas adequadas para que se obtenham os produtos. Os métodos computacionais quânticos e de docking molecular são os primeiros passos para o esclarecimento de mecanismos enzimáticos como os envolvidos na concepção de fármacos.
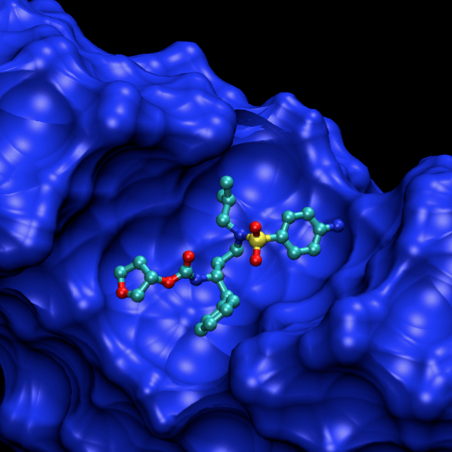
Por exemplo, o vírus HIV-1 da SIDA tem uma actividade enzimática de extrema complexidade. Produz três enzimas: a integrase que comanda a integração do seu material genético nas células infectadas, a transcriptase reversa, que converte o ARN viral de cadeia simples em ADN de cadeia dupla, pronto para ser integrado no ADN humano, e a protease que activa a destruição de proteínas sintetizadas nas células. A concepção de inibidores (fig. 5) que combatam esta doença tem tido conribuições do grupo de Maria João Ramos, das quais alguns aspectos, ilustrados com filmes, estão disponíveis em [22].
Figura 5. Um passo da associação de uma molécula com interesse terapêutico à protease do HIV-1
Outros aspectos computacionais no contexto da Bioquímica, e da Química Medicinal e Farmacêutica, também têm sido desenvolvidos por outros grupos, por exemplo, os de Nuno Micaelo [23], Vitor Félix [24], Daniel dos Santos [25] e Nuno Palma [26, 27]
Usando computadores e outros instrumentos, ou mesmo uma simples folha de papel e lápis, os investigadores e técnicos determinam valores para diferentes propriedades de sistemas e processos. Ou seja, realizam sempre medições, cujas fiabilidade, correcta apresentação e comparações só podem garantir-se através da definição internacional de sistemas de unidades, da avaliação de incertezas e erros, e da calibração dos instrumentos utilizados. Por outro lado, a saúde, segurança pública e ambiente exigem normas estritas que regulem as calibrações instrumentais, formulação de produtos e processos industriais. O incumprimento dessas normas na calibração dos altímetros de aviões e na formulação de fármacos ou pesticidas, por exemplo, é desastroso como, infelizmente e por vezes, se tem verificado.
Estes problemas, de qualquer campo da ciência e tecnologia, são do domínio da Metrologia (não confundir com Meteorologia) que utiliza, em particular, métodos computacionais da Quimiometria.
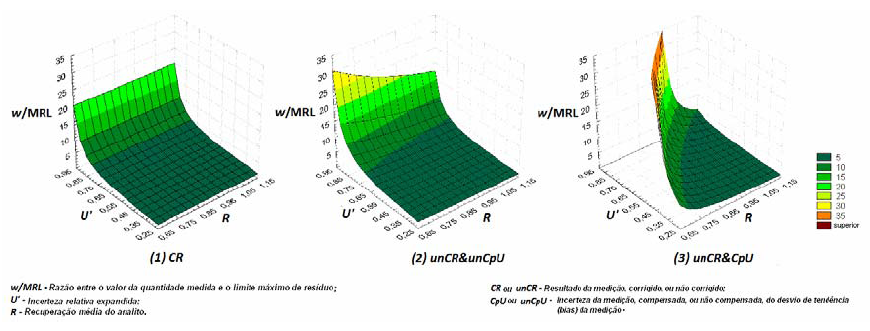
A descrição e a quantificação das espécies químicas envolvidas em equilíbrios químicos múltiplos e a modelação do desempenho metrológico de medições químicas capazes de identificar a origem de divergências interlaboratoriais, na avaliação da conformidade dos alimentos em matrizes complexas (fig. 6) com a legislação, são exemplos de situações com forte impacto socio-económico que beneficiam de ferramentas computacionais desenvolvidas ou aplicadas para o efeito. Alguns grupos, como o de Filomena Camões [28], têm-se dedicado a problemas destes domínio.
Figura 6. Modelo da compatibilidade de medições de resíduos de pesticidas em alimentos considerando diferentes convenções de apresentação dos resultados e estimativas da incerteza da medição.
No âmbito da metrologia de calibração instrumental, em particular, um outro grupo é o de Nieto de Castro [29].
Um texto de divulgação deve evitar, o mais possível, tecnicalidades mantendo, no entanto, o rigor científico. Além de uma digressão através dos objectivos, métodos e tipos de programas da Química Computacional, seleccionámos exemplos resumidos da actividade que se desenvolve em Portugal, representativos de algumas vertentes específicas.
Neste contexto, cabe-nos destacar os prémios Alberto Romão Dias e Vicente Seabra atribuídos, pela Sociedade Portuguesa de Química em Julho 2011, respectivamente a Maria José Calhorda [30] da Universidade de Lisboa, e a José Richard Gomes [31] da Universidade de Aveiro, em cujos grupos de investigação a Química Computacional desempenha um papel fundamental.
A divulgação científica é um dever cívico, especialmente dos cientistas. Creio que é uma das vias para estimular as gerações mais novas, combater a iliteracia científica e informar os meios sociais dos sucessos (e também dos insucessos, sublinhe-se) do constante esforço da Ciência em contribuir para o progresso e bem-estar das sociedades. Por muito sigilo que haja numa investigação, existem sempre maneiras de divulgar certos aspectos gerais e técnicas utilizadas, estas por vezes bem conhecidas, mantendo “o grau útil de ignorância” de outrem.
Termino recordando o que disse no início. Qualquer pessoa, curiosa e persistente, pode aprender Química Computacional servindo-se dum vulgar computador ligado à Internet. E, não é exagero afirmar, nem sempre é necessário ser-se especialista numa determinada área para produzir excelentes ideias científicas. Basta ler um pouco de História da Ciência para o confirmar. Aqui fica o desafio, caros leitores.
Aos colegas Pedro Fernandes (Fac. Ciências, Univ. Porto), João Aires de Sousa e Diogo Latino (Fac. Ciências e Tecn., Univ. Nova de Lisboa), Filomena Camões, Ricardo Bettencourt, Miguel Machuqueiro e Pedro Rodrigues (Fac. Ciências, Univ. Lisboa) pela disponibilização de algumas figuras e filmes, bem como comentários e sugestões sobre o texto. Ao avaliador deste artigo pela crítica e oportunas sugestões.
[1] Open Source
Physics
(http://www.opensourcephysics.org/) [3] "Using Artificial Intelligence in Chemistry and Biology, A Practical Guide", Hugh Cartwright, CRC Press, 2008.
[4] "Cinquentenário
da Simulação Computacional em Mecânica Estatística I. Os primeiros
passos", [5] "Cinquentenário da Simulação Computacional em Mecânica Estatística II. Desenvolvimento e aplicações fundamentais", F.Fernandes, Química, Bol. SPQ, 93 (2004) 49-60.
[6] "Monte
Carlo em Mecânica Estatística. Uma explicação simples com argumentos
cinéticos", [7] Pedro Rodrigues (http://webpages.fc.ul.pt/~fmfernandes/clusters_IYC2011/index_po.htm) [8] Rui Fartaria (https://uk.linkedin.com/in/rui-fartaria-1861a021/pt) [9] CICECO (http://www.ciceco.ua.pt/) [10] Canongia Lopes (http://cqe.ist.utl.pt/personal_pages/pages/jose_nuno_lopes.php/) [11] Fernando Fernandes (http://webpages.fc.ul.pt/~fmfernandes) [12] Ferreira Gomes (http://www.fc.up.pt/pessoas/jfgomes) [13] António Varandas (http://www.uc.pt/uid/tcc) [14] Benedito Cabral (http://gfm.cii.fc.ul.pt/people/bjcabral/) [15] Prates Ramalho (http://www.uevora.pt/pessoas/(id)/4267) [16] Aires de Sousa (http://joao.airesdesousa.com) [17] SPINUS (http://www.nmrdb.org/new_predictor/index.shtml?v=v2.34.2) [18] Diogo Latino (https://pt.linkedin.com/in/diogo-latino-25a91242) [19] Miguel Machuqueiro (http://intheochem.fc.ul.pt/research/artwork.html) [20] Cláudio Soares (http://www.itqb.unl.pt/~claudio/) [21] António Baptista (http://www.itqb.unl.pt/~baptista/) [22] Maria João Ramos (http://www2.fc.up.pt/A-quimica-do-dia-a-dia/) [23] Nuno Micaelo (https://pt.linkedin.com/in/nunomicaelo) [24] Vitor Félix (http://molecular-modeling.dq.ua.pt/) [25] Daniel dos Santos (http://www.imed.ul.pt/portal/index.php?option=com_content&view=article&id=215&Itemid=80) [26] Nuno Palma (http://www.linkedin.com/in/nunopalma64) [27] Bial (http://www.bial.com/pt/) [28] Filomena Camões (http://www.fc.ul.pt/node/2135) [29] Nieto de Castro (http://www.fc.ul.pt/node/2087) [30] Maria José Calhorda (http://intheochem.fc.ul.pt/) [31] José Richard Gomes (http://sweet.ua.pt/~f3963/)
|